Apoptose
Pour l’article homonyme, voir Apoptose (projet musical).
L'apoptose (ou mort cellulaire programmée) est le processus par lequel des cellules déclenchent leur autodestruction en réponse à un signal. C'est l'une des voies possibles de la mort cellulaire, qui est physiologique, génétiquement programmée, nécessaire au développement et à la survie des organismes multicellulaires[1]. Son équilibre avec la prolifération cellulaire permet l'homéostasie tissulaire. Contrairement à la nécrose, elle ne provoque pas de réaction du système immunitaire : les membranes plasmiques ne sont pas détruites, du moins dans un premier temps, et la cellule émet des signaux (en particulier, elle expose sur le feuillet externe de sa membrane plasmique de la phosphatidylsérine, un phospholipide normalement constitutif de son feuillet interne) qui permettront sa phagocytose par les cellules macrophagiques et particulièrement les macrophages.

Découverte et étymologie
modifierL'apoptose (du grec : apo, « au loin » et ptosis, « chute ») a été mise en évidence en 1972 par John Kerr, Andrew Wyllie et Alastair Currie lors de l'étude de tissu par microscopie électronique[2].
Ils choisirent ce mot apoptosis pour décrire le phénomène de mort cellulaire naturelle. Ce mot provient d'une locution grecque évoquant la « chute des feuilles »[3]. Elle avait déjà été employée en médecine par Hippocrate de Kos (460-377 av. J.-C.) dans son traité Des Instruments de réduction pour décrire la décomposition des tissus après la mort (« chute des os »)[3],[4].
Exemples de rôles physiologiques
modifierMorphogenèse
modifierL'apoptose joue un rôle dans la formation du corps d'un organisme, par exemple l'émergence des doigts. Au début de sa formation, la main ressemble à une moufle (ou une palme), puis les cellules se trouvant entre les futurs doigts disparaissent. De même, la disparition de l'appendice caudal, chez le fœtus humain, est due à ce phénomène d'apoptose. La régression de la queue chez les têtards lors de leur métamorphose en grenouille est elle aussi due à l'apoptose.
Elle joue un rôle dans la formation du cerveau : très tôt dans l'embryogenèse, le cerveau subit une vague apoptotique qui le remodèle. Ensuite, les neurones forment entre eux des liaisons synaptiques au hasard [réf. nécessaire], et une deuxième vague apoptotique élimine ceux qui n'ont pas établi de liaisons utiles.
Elle peut également avoir un rôle moteur, la rétraction des cellules mortes entraînant la mobilisation des tissus voisins. Ce mécanisme a notamment été décrit chez l'embryon de la drosophile[5].
Système immunitaire
modifierEn réponse à l'apparition d'un antigène étranger dans le corps, des lymphocytes B se mettent à produire chacun un anticorps particulier, en recombinant au hasard leurs gènes d'immunoglobulines (recombinaison VDJ). Ceux qui produisent des anticorps inactifs ou autoimmuns sont éliminés par apoptose. En cas d'infection virale, les lymphocytes T cytotoxiques produisent des molécules toxiques pour les cellules infectées ; ils sont détruits par apoptose lorsque l'infection est maîtrisée. C'est le cas dans l'infection par le VIH, mais cette voie entraîne par la suite des effets néfastes car les LT4 détruits en grande quantité ne peuvent plus sécréter les interleukines à l'origine de la sélection et de l'amplification clonale.
Différenciation intestinale
modifierLes cellules intestinales sont en perpétuel renouvellement (avec une durée de vie de quelques jours seulement) et migrent du bas des cryptes vers le sommet des villosités intestinales de l'intestin grêle où elles assurent leur fonction d'absorption des nutriments. Ultimement elles se décrochent et enclenchent un phénomène apoptotique particulier appelé anoïkose, dû à la perte de contact cellule-cellule ou cellule-matrice extracellulaire.

Dédifférenciation mammaire
modifierLes acini des glandes mammaires après la période d'allaitement, et en absence du maintien du signal de prolifération/différenciation dû à la prolactine, vont se résorber, et perdre un nombre important de cellules épithéliales par un processus d'apoptose.
Causes
modifier- Une cellule normale a constamment besoin que le corps lui confirme son utilité, aux moyens de facteurs de croissance. La perte de ces signaux peut déclencher un processus apoptotique.
- Des signaux émis à la suite des dommages subis par l'ADN (par exemple à la suite d'une irradiation aux rayons UV ou aux rayons X) sont capables de déclencher l'apoptose : en effet c'est alors soit une cellule potentiellement cancéreuse, soit une cellule totalement dysfonctionnelle. Dans les deux cas, cette cellule doit être éliminée sans dommage pour le reste du tissu adjacent.
- Des signaux hormonaux, notamment par les glucocorticoïdes, peuvent déclencher l'apoptose. C'est un mécanisme important, de régulation du système immunitaire.
- Pression sur le réticulum endoplasmique : lorsqu'une cellule a un problème dans la conformation d'une protéine, qui aboutit à une accumulation de cette protéine dans le réticulum endoplasmique, elle peut entrer en apoptose.
- La perte des contacts entre certaines cellules, ou bien entre ces cellules et leur matrice extracellulaire environnante induit de façon extrêmement rapide un processus apoptotique appelé anoïkose.
- L'apoptose peut aussi être causée par la dégradation des télomères des chromosomes. Elle peut être inhibée par les télomérases. Ce processus est aussi appelé horloge biologique.

Manifestation cytologique
modifierL'apoptose se manifeste sur ces cellules isolées (non regroupées). On constate sur les cellules concernées une compaction et une marginalisation de la chromatine nucléaire (pycnose) ainsi qu'une convolution des membranes cytoplasmiques et nucléaires et une condensation du cytoplasme.
L'intégrité des membranes est conservée au cours du processus apoptotique évitant ainsi toute réaction inflammatoire. Les corps apoptotiques sont protégés par une enveloppe membranaire issue de la convolution des membranes.
Étape 1 : purge de la membrane apoptotique
modifierLors de l'induction de l'apoptose par voie extrinsèque (activée par les récepteurs transmembranaires) ou intrinsèque (par l'intermédiaire des mitochondries)[7], la cellule en train de mourir peut générer des renflements ou des blebs ou bulles à la surface de la cellule, un processus connu sous le nom de claquage d'une membrane apoptotique[7]. La purge membranaire apoptotique est considérée comme la première étape (étape 1) du désassemblage des cellules apoptotiques, qui peut apparaître sous la forme de petites bulles superficielles aux premiers stades de l'apoptose ou de grandes bulles dynamiques membranaires aux étapes ultérieures[8],[9]. La formation de grandes bulles de membrane dynamiques pourrait faciliter la fragmentation des organites tels que le noyau au cours de la progression de l'apoptose[8],[10]. Le saignement des membranes apoptotiques est régulé par un certain nombre de facteurs moléculaires, en particulier le facteur protéine kinase 1 associée à la famille Rho des petites GTPases contenant un enroulement hélicoïdal (ROCK1 (en)) activée par la caspase[11],[12].
Étape 2 : formation de protubérances membranaires apoptotiques
modifierAprès le débordement de la membrane apoptotique, une cellule peut subir d'autres modifications morphologiques pour générer une variété de saillies minces de la membrane apoptotique, notamment des pointes de microtubules, des « apoptopodes » et des « apoptopodes en perles »[13],[14],[15]. La formation de ces saillies membranaires apoptotiques dépend souvent du type de cellule et représente la deuxième étape (étape 2) du désassemblage des cellules apoptotiques[8],[9] (figure 1). Par exemple, des pointes de microtubules ont été observées sur des cellules épithéliales squameuses apoptotiques[13]. Mécaniquement, la formation de pointes de microtubules dépend de la polymérisation des microtubules et de l'établissement du réseau de microtubules[13]. La formation de pointes de microtubules se réalise pour faciliter la séparation des bulles de membrane, ainsi que la distribution du contenu nucléaire dans les bulles de membrane[13]. Plus récemment, un autre type de saillie de la membrane apoptotique moins rigide et semblable à des cordes, appelée « apoptopodes » (« pieds de la mort »), a été identifié sur les cellules T, les thymocytes et les fibroblastes apoptotiques[14]. À l'instar des pointes de microtubules, la formation d'apoptopodes peut induire une séparation des blebs de la membrane[14]. En outre, les monocytes apoptotiques peuvent générer un autre type de saillie de la membrane apoptotique, appelée apoptopode perlée, qui ressemble à une « perle sur chaîne »[15]. La formation d'apoptopodes perlés commence par la génération et l'allongement d'une saillie semblable à l'apoptopode, qui se segmente et se présente sous la forme d'une chaîne de perles[15]. Actuellement[Quand ?], le seul régulateur moléculaire connu de la formation d'apoptopodes perlés et d'apoptopodes est le canal membranaire activé par la caspase PANX1 (en) (pannexine 1)[14],[15].
Étape 3 : fragmentation cellulaire
modifierEnfin, la libération de corps apoptotiques individuels liés à la membrane (généralement considérés comme ayant un diamètre d'environ 1 à 5 microns) représente l'étape finale (étape 3) du désassemblage des cellules apoptotiques[8],[9]. Bien que le mécanisme à la base du processus de fragmentation final ne soit pas bien défini, la dissociation des corps apoptotiques de différents types de saillies membranaires apoptotiques peut nécessiter une contrainte de cisaillement ou peut-être même une interaction avec les cellules voisines[8]. Il convient de noter qu’en plus des corps apoptotiques, des vésicules membranaires d’un diamètre inférieur à 1 micron sont également libérées au cours de l’apoptose[16].
La libération de corps apoptotiques à partir de la cellule en train de mourir a été proposée pour faciliter la communication entre cellules par le biais de protéines, de micro-ARN et d'ADN présents sur / dans des corps apoptotiques[17],[8],[18],[19]. Il a également été proposé que le désassemblage des cellules apoptotiques pourrait faciliter l'élimination des cellules mortes, car il pourrait s'avérer plus efficace pour une cellule phagocytaire d’engloutir des fragments cellulaires plus petits (c'est-à-dire des corps apoptotiques) plutôt qu'une cellule apoptotique comme un tout[8],[20].

Mécanismes moléculaires
modifier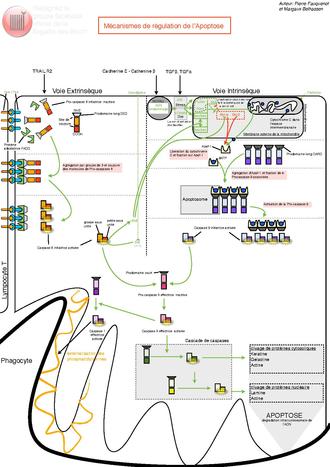
Le mécanisme d’apoptose est gouverné par deux voies principales d’activation :
- une voie dite extrinsèque, impliquant des récepteurs appartenant à la super-famille de protéines des récepteurs au facteur de nécrose tumorale (TNF de l'anglais : tumor necrosis factor) ;
- une voie dite intrinsèque mettant en jeu la mitochondrie ; cette voie est gouvernée principalement par des protéines appartenant à la super-famille de Bcl-2.

Ces deux voies conduisent à l’activation de protéases à cystéine (caspases) dites effectrices. Celles-ci sont responsables du clivage de plusieurs molécules, comme certaines protéines de structure, ce qui se traduit par des phénomènes morphologiques et biochimiques caractéristiques se terminant par le démantèlement de la cellule : exposition de phosphatidylsérine à la surface de la membrane cellulaire, arrêt de la réplication, fragmentation du noyau et du cytosquelette entraînant la formation de corps apoptotiques phagocytés par les cellules environnantes.
Voie extrinsèque
modifierComme son nom l'indique, la voie extrinsèque de l'apoptose est déclenchée par un signal extérieur à la cellule. Par exemple, cette voie est empruntée lorsqu'un lymphocyte T cytotoxique déclenche la mort d'une cellule indésirable. Dans ce cas, par un simple contact de membrane à membrane, la cellule immunitaire induit l'activation des récepteurs Fas et l'apoptose de la cellule ciblée. Les mécanismes induits par FasR peuvent être étendus à ceux d'autres récepteurs de mort tels que DR4/TRAIL-R1 et DR5/TRAIL-R2. L'activation de FasR par son ligand induit le recrutement d'un complexe appelé DISC (Death-Inducing Signaling Complex) composé de molécules adaptatrices FADD (Fas Associated Death Domain) et des procaspases initiatrices -8 et -10. FADD se lie par l'intermédiaire de son propre domaine de mort (Death Domain ou DD) aux DD des récepteurs Fas par des liaisons homotypiques électrostatiques[21]. FADD contient également un domaine effecteur de mort (Death Effector Domain ou DED) qui, par des liaisons homotypiques hydrophobes[22], permet le recrutement des caspases initiatrices. La formation de ce complexe entraîne, par un effet de proximité, l'autoclivage des caspases -8 et -10 qui sont alors relarguées dans le cytosol sous une forme dimérique active. Cela permet l'activation séquentielle des caspases effectrices, parmi lesquelles la caspase-3. Les caspases initiatrices -8 et -10 peuvent activer la voie intrinsèque via le clivage de la protéine Bid et ainsi amplifier le signal apoptotique[23]. Dans le cas de cellules dites « de type 1 », le déclenchement de l'apoptose n'est pas dépendante de la voie intrinsèque. Inversement, dans les cellules dites « de type 2 », l'activation de la voie intrinsèque est nécessaire à l'apoptose et son blocage résulte en la résistance des cellules à Fas.
Voie intrinsèque
modifierPar opposition à la voie extrinsèque, la voie intrinsèque (ou voie mitochondriale) de l'apoptose est généralement induite par des signaux internes à la cellule. Par exemple, cette voie est déclenchée par l'activation de p53 lors des dommages importants à l'ADN. L'activation de cette voie repose principalement sur la formation de pores de transition de perméabilité dans la membrane des mitochondries par l'ouverture du PTPC (Permeability Transition Pore Complex), un complexe multiprotéique de la membrane interne mitochondriale. Ces pores sont des canaux oligo-protéiques constitués au niveau de la membrane externe par la porine (ou VDAC : Voltage Dependent Anion Channel) et sur la membrane interne par l'ANT (Adenine Nucléotide Translocator). Cette phase d'activation s'accompagne d'une diminution du potentiel transmembranaire mitochondrial (Delta Psi m), suivi du gonflement de la matrice mitochondriale, d'une interruption du métabolisme énergétique aérobique et d'un stress oxydatif. Selon les modèles, il y a rupture ou ouverture de la mitochondrie, ce qui permet la libération dans le cytosol de molécules pro-apoptotiques normalement mitochondriales, telles que le cytochrome c, Smac/DIABLO, OMI/HtrA2, l'endonucléase-G ainsi que l’Apoptosis Inducing Factor (AIF). La phase de perméabilisation de la membrane mitochondriale n'est pas encore pleinement comprise. Néanmoins, celle-ci est décrite pour être sous le contrôle d'interactions complexes et dynamiques entre les membres pro- et anti-apoptotiques de la famille de Bcl-2[24]. Les membres anti-apoptotiques (Bcl-2, Bcl-xL, Mcl-1) forment des hétérodimères avec les membres pro-apoptotiques Bax et Bak, contrebalancent leur activité et permettent la stabilité de la mitochondrie. Lorsque la mitochondrie est activée, par exemple lors d'une activation de FasR et par l'intermédiaire de Bid, celui-ci intéragit et active directement Bax et Bak. Cela induit la perte d'interaction entre Bax, Bak et les membres anti-apoptotiques de la famille de Bcl-2, leur oligomérisation, la formation des pores et le relarguage des protéines mitochondriales[25]. Une fois devenus cytosoliques, les facteurs pro-apoptotiques mitochondriaux vont avoir des cibles et des effets spécifiques. Le cytochrome c cytosolique s'associe à APAF1 et la procaspase-9 pour former un complexe nommé apoptosome. La caspase-9 est activée au sein de ce complexe et est capable dʼactiver à son tour les caspases effectrices comme la caspase-3[26]. La procaspase-9 a plus dʼaffinité pour lʼapoptosome que la caspase-9 active ce qui permet une rotation/activation des procaspases-9 au sein d'un même apoptosome[27]. Smac/DIABLO et OMI/HtrA2 ciblent et inhibent les protéines de la famille des IAP (Inhibitor of apoptosis) favorisant ainsi l'activité des caspases[28],[29]. Le facteur AIF est redistribué vers le cytosol puis vers le noyau cellulaire pour induire une mort par apoptose qui a la particularité d'être indépendante des caspases et ne nécessiter aucun autre intermédiaire[30].
Fragmentation de l'ADN
modifierDurant l’apoptose, l’ADN est digéré de façon très spécifique en fragments dont les tailles sont des multiples de 180 paires de bases, ce qui cause une distribution très caractéristique des fragments d’ADN en « échelle » lorsqu'ils sont séparés par électrophorèse suivant leur taille. Cette taille est révélatrice de l'espacement entre deux nucléosomes consécutifs. Cette digestion est assurée par les protéines CAD (Caspase Activated DNase), existant en temps normal sous forme inactive en association avec une ICAD (Inhibitor of Caspase Activated DNase). Cette ICAD cache la séquence NLS de CAD et le clivage de cette association par une caspase va permettre à la protéine CAD de rentrer dans le noyau, qui jouera son rôle de DNase clivant l'ADN. La fragmentation de l'ADN est utilisée pour détecter des cellules en apoptose dans un tissu grâce à la technique du TUNEL (Terminal Transferase dUTP Nick End Labeling)[31].
L'un des mécanismes de l'apoptose dans les cellules cardiaques post ischémiques semble consister en la nitration des protéines cellulaires par un excès de peroxynitrites[32]. Ces peroxynitrites induisent également l'apoptose des monocytes[33] et des lymphocytes T[34].
Par exemple quand les cellules ne reçoivent pas en permanence de leurs voisines des messages inhibant leur autodestruction, elles disparaissent spontanément.
Conséquences d'un dysfonctionnement de l'apoptose
modifierMaladies ou affections induites par le blocage de l'apoptose
modifierLes cellules cancéreuses sont généralement des cellules dans lesquelles ce mécanisme ne fonctionne plus. Elles survivent et se multiplient en dépit d'anomalies génétiques survenues au cours de la vie de la cellule, alors que normalement elles auraient dû être détruites par apoptose.
La réactivation du mécanisme d'apoptose a pu toutefois être obtenue chez des cellules cancéreuses de rat[35].
Certains pathogènes empêchent l'induction de l'apoptose, comme HHV8 (herpesvirus responsable du sarcome de Kaposi), qui code la protéine v-FLIP, empêchant l'apoptose induite par les récepteurs de mort.
Certaines maladies neurodégénératives comme les tauopathies, sont également des maladies où les mécanismes apoptotiques sont impliqués, conduisant à la survie de la protéine tau pathogène qui peut alors s'accumuler anormalement, jusqu'à la mort de la cellule nerveuse. C'est le cas de la paralysie supranucléaire progressive, de la maladie d'Alzheimer, etc.
Maladies ou affections causées par l’activation intempestive de l’apoptose
modifierDes recherches récentes[36] semblent montrer que le développement du sida en tant que maladie serait lié au déclenchement intempestif de l’apoptose des lymphocytes gérant la réponse immunitaire, ce qui permet le développement de maladies et infections opportunes. Cela ne remet pas en cause le rôle actif du virus VIH comme cause effective de cette maladie, bien que celui-ci soit bien détecté et tué par les lymphocytes.
Toutefois le blocage du virus par les anticorps produits par les lymphocytes conduirait le virus à produire avant sa destruction complète une réponse chimique de défense destinée à provoquer l’apoptose massive de tous les lymphocytes voisins, voire à faire fabriquer par les macrophages (qui absorberaient le virus neutralisé par les anticorps en même temps que le message chimique provoquant leur apoptose) cette réponse chimique qui provoquerait « à distance » le suicide de nombreux autres lymphocytes voisins alors même qu’ils n’ont jamais été directement en contact avec le VIH. En d’autres termes, le VIH provoquerait une réponse exacerbée du système immunitaire contre lui-même. C'est alors un effet « boule de neige », où un système morphologique est détourné de ses fonctions par une réponse non contrôlée, semblable à d’autres phénomènes auto-induits comme les allergies (elles aussi liées à un facteur déclenchant externe).
En provoquant cette réaction, une partie des copies du VIH parviendrait ainsi à échapper à l’action des lymphocytes T, dont un grand nombre se sont apoptosés après que quelques-uns d'entre eux seulement ont neutralisé de nombreux virus voisins. Cela expliquerait aussi pourquoi l'éradication totale du virus par le système immunitaire n’est pas possible sans une aide extérieure non sensible au phénomène de l’apoptose (une aide apportée par les médicaments anti-rétroviraux qui s'attaquent spécifiquement au VIH pour éviter que les lymphocytes s’en chargent en activant alors l'apoptose de leurs voisins par l'action ultérieure des macrophages éliminateurs)[37].
La compréhension des mécanismes chimiques de l'apoptose pourrait ainsi pallier cette fragilité intrinsèque du système immunitaire, et permettre donc le développement d'un type de vaccin particulier, non destiné à activer la réponse immunitaire contre le VIH (puisque celle-ci a bien lieu naturellement et produit de nombreux anticorps) mais à bloquer le déclenchement intempestif de l'apoptose des lymphocytes T CD4 chargés de leur neutralisation (et de la neutralisation des autres sources d'infection). Dans le cas du sida, on ne sait pas exactement quel lymphocyte possède cette fragilité (dangereuse uniquement pour les autres types de cellules mais pas lui-même directement), mais on peut penser qu'elle se situe au niveau des macrophages chargés d'éliminer les virus neutralisés par les anticorps.
Ce comportement intempestif des mêmes macrophages (les poubelles de l’organisme qui peuvent générer par leur action des tas de produits toxiques et agents chimiques difficiles à éliminer isolément) est également impliqué dans d'autres types de réactions exacerbées de l’organisme comme certaines allergies (où cette réaction, très largement autoentretenue, se fait à destination d'autres types de cellules que les lymphocytes immunitaires), et est soupçonné également dans d'autres types de maladies dégénératives (qui possèdent aussi un facteur déclenchant externe, pas nécessairement de nature infectieuse) ou certaines réactions exacerbées face à un stress (par exemple l'extension des brûlures).
L'apoptose chez les plantes à fleurs (Angiospermes)
modifierChez les angiospermes, l'apoptose est un processus majeur de l'immunité végétale, elle est observée en réponse aux stress abiotiques, et elle est considérée comme un élément clé du développement du gamétophyte[38].
Notes et références
modifier- Patrick Pla, « L'apoptose »
 , sur Biologie cellulaire et génétique du développement (consulté le )
, sur Biologie cellulaire et génétique du développement (consulté le )
- J.F.R. Kerr, A.H. Willie, A.R. Currie, « Apoptosis: a basic biological phenomenon with wide-ranging implications in tissue kinetics », Br J Cancer, no 26, 1972, p. 239-57 .
- chap 2, l'apoptose de la thèse de doctorat en sciences vétérinaires, Identification de nouveaux mécanismes régulateurs de l'apoptose des granulocytes, 2007.
- « Useful known and unknown views of the father of modern medicine, Hippocrates and his teacher Democritus », Hellenic journal of nuclear medicine, The Journal of the Hellenic Society of Nuclear Medicine, Hell J Nucl Med, 2008, vol. 11, no 1, p. 2-4.
- L.A. Davidson, « Apoptosis turbocharges epithelial morphogenesis », Science, no 21, 2008, p. 1641-1642.
- A. Smith, M. Parkes, G. Atkin-Smith, R. Tixeira, I. Poon, « Cell disassembly during apoptosis », WikiJournal of Medicine, vol. 4, no 1, 2017, p. 8. doi:10.15347/wjm/2017.008 (ISSN 2002-4436) [lire en ligne].
- S. Elmore, « Apoptosis: a review of programmed cell death », Toxicol. Pathol., vol. 35, no 4, , p. 495-516 (PMID 17562483, PMCID 2117903, DOI 10.1080/01926230701320337).
- G.K. Atkin-Smith, I.K. Poon, « Disassembly of the Dying: Mechanisms and Functions », Trends Cell Biol., vol. 27, no 2, , p. 151-62 (PMID 27647018, DOI 10.1016/j.tcb.2016.08.011).
- R. Tixeira, S. Caruso, S. Paone, A.A. Baxter, G.K. Atkin-Smith, M.D. Hulett, I.K. Poon, « Defining the morphologic features and products of cell disassembly during apoptosis », Apoptosis, vol. 22, no 3, , p. 475-7 (PMID 28102458, DOI 10.1007/s10495-017-1345-7).
- G. Wickman, L. Julian, M.F. Olson, « How apoptotic cells aid in the removal of their own cold dead bodies », Cell Death Differ., vol. 19, no 5, , p. 735-42 (PMID 22421963, PMCID 3321633, DOI 10.1038/cdd.2012.25).
- M.L. Coleman, E.A. Sahai, M. Yeo, M. Bosch, A. Dewar, M.F. Olson, « Membrane blebbing during apoptosis results from caspase-mediated activation of ROCK I », Nat. Cell Biol., vol. 3, no 4, , p. 339-45 (PMID 11283606, DOI 10.1038/35070009).
- M. Sebbagh, C. Renvoizé, J. Hamelin, N. Riché, J. Bertoglio, J. Bréard, « Caspase-3-mediated cleavage of ROCK I induces MLC phosphorylation and apoptotic membrane blebbing », Nat. Cell Biol., vol. 3, no 4, , p. 346-52 (PMID 11283607, DOI 10.1038/35070019).
- D.K. Moss, V.M. Betin, S.D. Malesinski, J.D. Lane, « A novel role for microtubules in apoptotic chromatin dynamics and cellular fragmentation », J. Cell. Sci., vol. 119, no Pt 11, , p. 2362-74 (PMID 16723742, PMCID 1592606, DOI 10.1242/jcs.02959).
- I.K. Poon, Y.H. Chiu, A.J. Armstrong, J.M. Kinchen, I.J. Juncadella, D.A. Bayliss, K.S. Ravichandran, « Unexpected link between an antibiotic, pannexin channels and apoptosis », Nature, vol. 507, no 7492, , p. 329-34 (PMID 24646995, PMCID 4078991, DOI 10.1038/nature13147).
- G.K. Atkin-Smith, R. Tixeira, S. Paone, S. Mathivanan, C. Collins, M. Liem, K.J. Goodall, K.S. Ravichandran, M.D. Hulett, I.K. Poon, « A novel mechanism of generating extracellular vesicles during apoptosis via a beads-on-a-string membrane structure », Nat. Commun., vol. 6, , p. 7439 (PMID 26074490, PMCID 4490561, DOI 10.1038/ncomms8439).
- C. Lynch, M. Panagopoulou, C.D. Gregory, « Extracellular Vesicles Arising from Apoptotic Cells in Tumors: Roles in Cancer Pathogenesis and Potential Clinical Applications », Front. Immunol., vol. 8, , p. 1174 (PMID 29018443, PMCID 5614926, DOI 10.3389/fimmu.2017.01174).
- Y. Berda-Haddad, S. Robert, P. Salers, L. Zekraoui, C. Farnarier, C.A. Dinarello, F. Dignat-George, G. Kaplanski, « Sterile inflammation of endothelial cell-derived apoptotic bodies is mediated by interleukin-1α », Proc. Natl. Acad. Sci. U.S.A., vol. 108, no 51, , p. 20684-9 (PMID 22143786, PMCID 3251090, DOI 10.1073/pnas.1116848108).
- .
- A. Zernecke, K. Bidzhekov, H. Noels, E. Shagdarsuren, L. Gan, B. Denecke, M. Hristov, T. Köppel, M.N. Jahantigh, E. Lutgens, S. Wang, E.N. Olson, A. Schober, C. Weber, « Delivery of microRNA-126 by apoptotic bodies induces CXCL12-dependent vascular protection », Sci. Signal., vol. 2, no 100, , ra81 (PMID 19996457, DOI 10.1126/scisignal.2000610).
- E. Witasp, W. Uthaisang, C. Elenström-Magnusson, R. Hanayama, M. Tanaka, S. Nagata, S. Orrenius, B. Fadeel, « Bridge over troubled water: milk fat globule epidermal growth factor 8 promotes human monocyte-derived macrophage clearance of non-blebbing phosphatidylserine-positive target cells », Cell Death Differ., vol. 14, no 5, , p. 1063-5 (PMID 17256011, DOI 10.1038/sj.cdd.4402096).
- E.J. Jeong, S. Bang, T.H. Lee, Y.I. Park, W.S. Sim, K.S. Kim, « The solution structure of FADD death domain. Structural basis of death domain interactions of Fas and FADD », J Biol Chem., vol. 274, no 23, 4 juin 1999, p. 16337-42.
- M. Eberstadt, B. Huang, Z. Chen, R.P. Meadows, S.C. Ng, L. Zheng, M.J. Lenardo, S.W. Fesik, « NMR structure and mutagenesis of the FADD (Mort1) death-effector domain », Nature, vol. 392, no 6679, 30 avril 1998, p. 941-5.
- H. Yamada, S. Tada-Oikawa, A. Uchida, S. Kawanishi, « TRAIL causes cleavage of bid by caspase-8 and loss of mitochondrial membrane potential resulting in apoptosis in BJAB cells », Biochem Biophys Res Commun., vol. 265, no 1, novembre 1999, p. 130-3.
- N. Volkmann, F.M. Marassi, D.D. Newmeyer, D. Hanein, « The rheostat in the membrane: BCL-2 family proteins and apoptosis », Cell Death Differ., vol. 21, no 2, février 2014, p. 206-15. doi: 10.1038/cdd.2013.153. Epub 25 octobre 2013.
- R. Eskes, S. Desagher, B. Antonsson, J.C. Martinou, « Bid induces the oligomerization and insertion of Bax into the outer mitochondrial membrane », Mol. Cell. Biol., vol. 20, no 3, février 2000, p. 929-35.
- P. Li, D. Nijhawan, I. Budihardjo, S.M. Srinivasula, M. Ahmad, E.S. Alnemri, X. Wang, « Cytochrome c and dATP-dependent formation of Apaf-1/caspase-9 complex initiates an apoptotic protease cascade », Cell., vol. 91, no 4, 14 novembre 1997, p. 479-89.
- S. Malladi, M. Challa-Malladi, H.O. Fearnhead, S.B. Bratton, « The Apaf-1*procaspase-9 apoptosome complex functions as a proteolytic-based molecular timer », EMBO J., vol. 28, no 13, 8 juillet 2009, p. 1916-25. doi: 10.1038/emboj.2009.152. Epub 4 juin 2009.
- A.M. Verhagen, P.G. Ekert, M. Pakusch, J. Silke, L.M. Connolly, G.E. Reid, R.L. Moritz, R.J. Simpson, D.L. Vaux, « Identification of DIABLO, a mammalian protein that promotes apoptosis by binding to andantagonizing IAP proteins », Cell., vol. 102, no 1, 7 juillet 2000, p. 43-53.
- A.M. Verhagen, J. Silke, P.G. Ekert, M. Pakusch, H. Kaufmann, L.M. Connolly, C.L. Day, A. Tikoo, R. Burke, C. Wrobel, R.L. Moritz, R.J. Simpson, D.L. Vaux, « HtrA2 promotes cell death through its serine protease activity and its ability to antagonize inhibitor of apoptosis proteins », J Biol Chem., vol. 277, no 1, 4 janvier 2002, p. 445-54. Epub 16 octobre 2001.
- S.P. Cregan, V.L. Dawson, R.S. Slack, « Role of AIF in caspase-dependent and caspase-independent cell death », Oncogene, vol. 23, no 16) 12 avril 2004, p. 2785-96.
- http://www.protocol-online.org/prot/Cell_Biology/Apoptosis/Terminal_Transferase_dUTP_Nick_End_Labeling__TUNEL__Assay/.
- Academic Emergency Medicine - Wiley Online Library.
- [PDF]« Contrasting effects of NO and peroxynitrites on HSP70 expression and apoptosis in human monocytes ».
- « Functional studies of an HIV-1 encoded glutathione… », Biofactors, 2006 - PubMed result.
- A. Athanasiou, P.A. Smith, S. Vakilpour, et al., « Vanilloid receptor agonists and antagonists are mitochondrial inhibitors: how vanilloids cause non-vanilloid receptor mediated cell death », Biochem. Biophys. Res. Commun., vol. 354, no 1, 2007, p. 50-5. doi:10.1016/j.bbrc.2006.12.179 .
- André Eyquem, Joseph E. Alouf et Luc Montagnier, Traité de microbiologie clinique : troisièmes mises à jour et compléments, , 107 p. (ISBN 978-88-299-1675-7, lire en ligne), p. 47.
- http://www.pasteur.fr/actu/presse/com/dossiers/Sida/recherchesIPParis.htm.
- (en) « Only in dying, life: programmed cell death during plant development », Trends in Plant Science, vol. 20, no 2, , p. 102-113 (ISSN 1360-1385, DOI 10.1016/j.tplants.2014.10.003, lire en ligne, consulté le ).
Annexes
modifierBibliographie
modifier
Certaines informations figurant dans cet article ou cette section devraient être mieux reliées aux sources mentionnées dans les sections « Bibliographie », « Sources » ou « Liens externes » ().
Vous pouvez améliorer la vérifiabilité en associant ces informations à des références à l'aide d'appels de notes.
- Jean-Claude Ameisen, La Sculpture du vivant : Le suicide cellulaire ou la mort créatrice, Point Seuil, 2003.
- Ph.J. Coulomb et J.M. Lacombe, « Le brunissement des olives », PDF de la Faculté des Sciences d’Avignon, , p. 10 (lire en ligne).
Articles connexes
modifierLiens externes
modifier- Ressources relatives à la santé :
- Cell Death & Differentiation : la revue scientifique internationale sur l'apoptose.
- « Apoptose : mourir peut attendre », La Science, CQFD, France Culture, 20 septembre 2022.